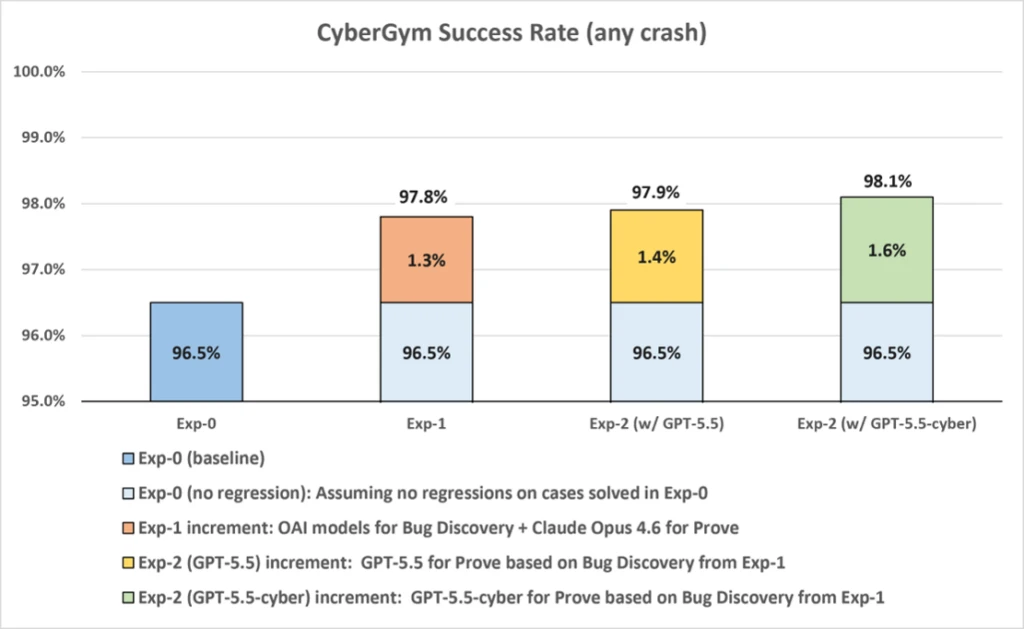
What Happened
Microsoft Research has developed an artificial intelligence method that can convert standard, hematoxylin and eosin (H&E)-stained pathology slides into detailed spatial proteomics maps. This process, detailed in a research paper, aims to replicate data traditionally obtained through costly and complex technologies like multiplexed immunofluorescence (mIF) or imaging mass cytometry.
The core achievement is the generation of spatially resolved protein expression profiles from a single, widely available H&E slide. This is significant because spatial proteomics provides a map of where specific proteins are expressed within a tissue sample, offering crucial biological and clinical insights, particularly in cancer research. However, the specialized equipment and reagents required are expensive, have limited throughput, and are not universally available.
How It Works (The Technical Leap)
The AI model is trained to learn the mapping between the visual patterns in an H&E image and the corresponding protein expression patterns that would be revealed by a spatial proteomics assay. H&E staining highlights cellular and tissue structures (nuclei in blue, cytoplasm and extracellular matrix in pink), but does not tag specific proteins.
The research likely involves a deep learning architecture, such as a conditional generative adversarial network (cGAN) or a vision transformer, trained on paired datasets. These datasets would consist of:
- The Input: A digitized H&E whole-slide image (WSI).
- The Target Output: The corresponding spatial proteomics map (e.g., from mIF) for the same tissue region, showing the expression levels and locations of multiple proteins.
By learning from these pairs, the model infers the complex, non-linear relationships between tissue morphology and protein expression. Once trained, it can predict a proteomic map for a new H&E slide without ever needing to run the physical proteomics assay.
Context & Implications
This work sits at the intersection of computational pathology and AI-powered biomarker discovery. The standard H&E slide is the most common and fundamental diagnostic tool in pathology. Extracting vastly more molecular information from it represents a major efficiency gain.
The primary claimed benefits are:
- Accelerated Analysis: The AI inference can generate a proteomic map in minutes from an existing digital slide, bypassing a lab process that can take days.
- Lowered Costs: It eliminates the need for expensive antibodies, fluorescent tags, and specialized imaging hardware for initial proteomic profiling.
- Expanded Access: Any clinic or research lab with a standard slide scanner and computational resources could, in principle, generate spatial proteomics data, democratizing access to this high-resolution biological data.
In practice, this could allow retrospective analysis of vast archives of H&E slides, unlocking new biomarkers from historical cancer cohorts. It could also serve as a triage tool, helping pathologists decide which cases truly warrant running a full, physical multiplex assay.
What the Source Doesn't Tell Us (Key Open Questions)
The source tweet and linked announcement are high-level. Critical technical details for evaluation are missing:
Model Performance: What is the fidelity of the predicted protein maps? Quantitative metrics (e.g., Pearson correlation, structural similarity index) comparing AI-generated maps to ground-truth experimental maps are essential.
Validation Scope: On which cancer types and proteins was the model validated? Performance likely varies across tissue and protein types.
Architecture & Data: The specific model architecture, training dataset size, and computational requirements are not specified.
Clinical Correlation: The ultimate test is whether AI-derived proteomic features are as predictive of patient outcomes or treatment response as those from physical assays. This clinical validation is a separate, significant step.
While the promise is substantial, the impact hinges on these unpublished technical validations. The approach is part of a growing trend of using AI to "translate" between biomedical data modalities, but its adoption will depend on proven accuracy and robustness.