In the race to develop artificial intelligence for scientific discovery, bigger has often been considered better. The prevailing wisdom in AI research has emphasized scaling—more parameters, more data, more computational power. But a groundbreaking new approach from researchers challenges this paradigm, demonstrating that sometimes, smarter training beats sheer size.
Published on arXiv on February 10, 2026, the paper "RxnNano: Training Compact LLMs for Chemical Reaction and Retrosynthesis Prediction via Hierarchical Curriculum Learning" introduces a remarkably efficient AI model that outperforms much larger systems in predicting chemical reactions—a critical task for accelerating drug discovery and materials science.
The Scaling Problem in Chemical AI
Chemical reaction prediction involves determining the products of a chemical reaction given specific reactants and conditions, while retrosynthesis works backward from a target molecule to identify possible synthetic pathways. Both are fundamental to pharmaceutical development, where identifying viable synthesis routes can mean the difference between a promising drug candidate reaching clinical trials or remaining stuck in the lab.
Current AI approaches have largely followed the scaling trend prevalent in general large language models (LLMs). Researchers have fine-tuned massive models with billions of parameters on chemical datasets, assuming that more capacity would naturally lead to better chemical understanding. However, this approach has limitations. As the RxnNano researchers note, these methods often "bypass fundamental challenges in reaction representation and fail to capture deep chemical intuition like reaction common sense and topological atom mapping logic."
In essence, bigger models trained on more data don't necessarily develop true chemical understanding—they may simply memorize patterns without grasping the underlying principles that govern chemical transformations.
A New Framework: Quality Over Quantity
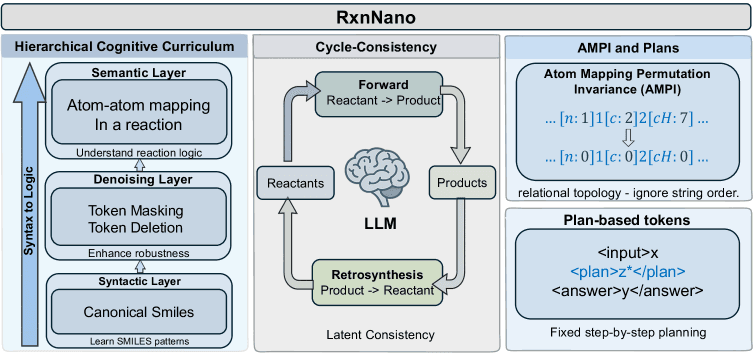
The RxnNano team proposes a radical alternative: instead of scaling up, they focus on instilling genuine chemical knowledge through innovative training techniques. Their compact 0.5-billion-parameter model achieves what models ten times larger cannot, thanks to three key innovations:
1. Latent Chemical Consistency
This objective models chemical reactions as movements on a continuous chemical manifold—a mathematical space where similar chemicals are located near each other. By ensuring transformations are reversible and physically plausible within this space, the model learns the fundamental constraints of chemistry rather than just statistical patterns in reaction data.
2. Hierarchical Cognitive Curriculum
Inspired by how humans learn complex subjects, this approach trains the model through progressive stages. It begins with mastering the "syntax" of chemical notation (how to properly represent molecules and reactions), then progresses to "semantic" reasoning about chemical properties and interactions, and finally advances to strategic planning for complex multi-step syntheses.
3. Atom-Map Permutation Invariance (AMPI)
Chemical reactions can be represented in multiple equivalent ways depending on how atoms are numbered. AMPI forces the model to recognize that these different representations describe the same underlying reaction, teaching it to focus on the invariant relational topology—the actual connections between atoms that change during reactions.
Performance That Defies Expectations
The results are striking. RxnNano achieves a 23.5% improvement in Top-1 accuracy on rigorous chemical prediction benchmarks compared to existing methods, all without test-time augmentation techniques that artificially boost performance. It outperforms not only specialized chemical AI systems but also fine-tuned general LLMs with more than 7 billion parameters.
This performance gap is particularly significant because chemical reaction prediction has traditionally been considered a "hard" problem for AI—one requiring deep domain knowledge rather than just pattern recognition. The fact that a relatively small model can excel where larger ones struggle suggests that the research community may have been overlooking crucial aspects of how to teach AI systems scientific reasoning.
Implications for AI and Scientific Discovery
The success of RxnNano has several important implications:
For Drug Discovery: Faster, more accurate reaction prediction could significantly accelerate pharmaceutical research. Identifying synthesis pathways is often a bottleneck in drug development, and improved AI tools could help bring promising treatments to market more quickly.
For AI Research: The work challenges the prevailing "scale is all" mentality in AI development. It demonstrates that for specialized domains like chemistry, carefully designed training objectives and curricula can achieve better results than simply throwing more parameters and data at a problem.
For Scientific AI More Broadly: The principles behind RxnNano—focusing on instilling domain knowledge, using progressive learning curricula, and designing invariance properties—could be applied to other scientific fields where AI is increasingly used, from materials science to climate modeling.
For Computational Efficiency: At just 0.5 billion parameters, RxnNano is far more accessible than multi-billion parameter models. Researchers and organizations with limited computational resources could deploy such models more easily, potentially democratizing access to advanced chemical prediction tools.
Looking Ahead
The researchers have made RxnNano available on GitHub, encouraging further development and application. As AI continues to transform scientific research, approaches like RxnNano's that prioritize understanding over scale may point the way toward more efficient, effective, and interpretable scientific AI systems.
What makes this development particularly exciting is its timing. As concerns grow about the environmental impact and computational costs of ever-larger AI models, RxnNano demonstrates that alternative paths exist—paths that might lead not only to more sustainable AI but to systems that actually understand the domains they're designed to help explore.
In the quest to harness AI for scientific breakthrough, sometimes thinking small—or at least, thinking smarter about how we train—might be the biggest leap forward.